Knowing where a protein is localized in cells is important in understanding its functions, in identifying the organelles that it helps to regulate, and in determining its binding partners. Proteins have been localized to cellular organelles by subcellular fractionation, which typically involves homogenizing the cells and then separating the organelles from each other on sucrose gradients. This is a good method for isolating sarcoplasmic reticulum (SR) from skeletal muscle, and we have used it extensively, but it isn’t very useful for showing that particular proteins are in the sarcolemma or the transverse tubules, or if they concentrate at the neuromuscular junction. We have turned to fluorescence methods to address these questions.
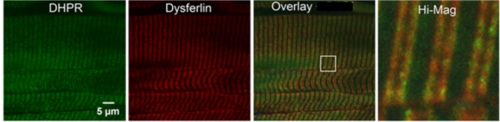
One method for localizing proteins within cells is by immunofluorescence. For this method, cells are first fixed and permeabilized with a water-soluble solvent (typically ethanol at -20oC) or fixed with paraformaldehye and permeabilized with a mild detergent. Cells are then incubated with a rabbit or mouse antibody specific for the protein of interest, followed by a secondary antibody (e.g., goat anti-mouse IgG) covalently tagged with a fluorescent molecule, such as fluorescein. The source of the fluorescence within the cells can be identified and compared to known markers of the organelles (SR, t-tubules, etc.) to identify the compartment in which the protein of interest is localized. This is most easily done by double labeling, using a different species of antibody to the marker protein and a secondary specific for that species that is tagged with a different fluorophore. This method localizes the proteins natural to the cell, called “endogenous proteins.” An example of near complete colocalization is shown in the “Spectrins at the Neuromuscular Junction” section. An example of only partial colocalization of dysferlin with the L-type Ca channel (DHPR) is shown below. The white box in the 3rd panel is expanded for a high magnification view to the right). Yellow indicates colocalization.
A second method for localizing proteins within cells is by creating a chimeric version of the protein, in which the DNA encoding the protein (or a fragment of it) is linked in frame to DNA encoding a fluorescent protein, such as Green fluorescent protein or Red fluorescent protein. After transfection into muscle cells, the chimeric DNA encodes a chimeric protein that fluoresces. As this is encoded by a synthetic DNA and is therefore distinct from the endogenous protein, it is called “exogenous.” The source of the fluorescence indicates the location of the exogenous, chimeric protein in the cells. The figure below shows sAnk1 coupled to an mCherry (red) fluorescent protein, compared to mCherry alone, after expression in a myofiber.

Good localization studies often require careful comparison of results with exogenous and endogenous versions of the protein of interest. They also require the highest possible optical resolution available. To achieve this, we employ different kinds of “confocal” microscopes, which use lasers as sources of the light to excite the fluorescent antibodies or chimeric proteins, and which limit the optical planes from which the fluorescent light emitted by these proteins can be detected. Without confocal optics, optimal resolution is ~0.25 µm in the X-Y axis, and ~2 µm in the Z-axis; with a confocal microscope, these values are ~0.13 µm in the X-Y axis, and ~0.75 µm in the Z-axis. (NB: The resolution of a light microscope, including those employing confocal methods, is limited by the numerical aperture of the lenses and the wavelength of the light beam). We are currently experimenting with state-of-the-art methods, including SIM (Structured Illumination Microscopy), and FRET (Förster Resonance Energy Transfer), which, coupled with confocal methods, can further increase the resolution ~2 fold (SIM) and can co-localize pairs of molecules within ~10 nm of each other (FRET).