Organization and Stability of Intracellular Membranes
We have been studying the organization of the sarcoplasmic reticulum (SR) and transverse tubule (TT) systems of skeletal muscle, to learn how they form, how they are compartmentalized, and what stabilizes them against potential damage caused by the changes in shape associated with muscle contraction and relaxation. Our work on TT focuses on dysferlin, the protein that is missing in patients with Limb Girdle Muscular Dystrophy Type 2B (LGMD2B) and Miyoshi Myopathy (MM). Our work on SR focuses on an unusually small form of ankyrin, which we call small ankyrin 1 (sAnk1; others call it Ank1.5) and its role in the network compartment of the SR.
Small Ankyrins and the Sarcoplasmic Reticulum
Our studies on sAnk1 have shown that it is an integral membrane protein containing two ankyrin repeat domains (shown below, modeled vs 2 ankyrin repeats of Notch 1 on the left, and in a space filling model on the right) that is enriched in the compartment of the SR called the “network SR” (nSR), which surrounds the middle and the ends of each contractile unit, or sarcomere. Knock-down of sAnk1 with siRNA destabilizes the nSR and significantly reducing its ability to take up and store Ca2+. Mutagenesis of the transmembrane domain of sAnk1 results in its mislocalization and accumulation in an inappropriate membrane compartment, without SERCA (the Ca2+-ATPase responsible for sequestering Ca2+ in the SR lumen). This also reduces Ca2+ stores in the SR lumen, which we measure directly with a fluorescent Ca2+-sensitive dye, using methods we developed in our laboratory. Thus, sAnk1 is needed for normal SR organization and probably needs to be close to SERCA in the SR membrane for effective Ca2+ accumulation. We are currently testing the hypothesis that sAnk1 forms a molecular complex with SERCA, as do sarcolipin and phospholamban, two other small integral membrane proteins of the SR, but that unlike these two proteins, which are inhibitory, sAnk1 promotes SERCA activity.

We have also been studying the factors that determine the localization of sAnk1 at two distinct and relatively distant (~1 µm) regions of the nSR, the region over the middle of the sarcomere, and the region over the ends. Our studies have identified a set of amino acids on one surface of the ankyrin-like repeat domains of sAnk1 (figure above) that mediate its ability to bind to a large protein, called “obscurin”, that concentrates at the edge of the contractile apparatus around the middle of each sarcomere. When we mutagenesize these residues on sAnk1 and introduce the mutated protein into skeletal myofibers, the mutant sAnk1 fails to accumulate around the middle of the sarcomere but it still accumulates around the ends. We are currently using additional mutagenesis to test the idea that sAnk1 accumulates around the sarcomere ends through its ability to interact with the N-terminal region of titin. These studies therefore have the potential of revealing important mechanisms responsible for the organization and stability of the nSR, which is itself essential for normal muscle function.
Finally, we have examined the interactions of sAnk1 with SERCA and find that these two proteins interacts directly, and that binding is mediated by both the cytoplasmic and transmembrane domains of sAnk1. Their interaction results in mild inhibition of SERCA activity, but protection of SERCA activity from further inhibition by sarcolipin. A model of the complex formed by SERCA (in white) and the transmembrane domain of sAnk1 (in green) is shown below.

Dysferlin and the Transverse Tubules
Our studies of dysferlin focus on its role in TT. Although many laboratories have previously attributed a role for dysferlin at the sarcolemma, we have shown unequivocally that it concentrates selectively in the TT (see below and Kerr et al., 2013) and that in its absence, TT are destabilized leading to the development of muscular dystrophy (At least two forms of dystrophy, Limb Girdle Muscular Dystrophy type 2B and Miyoshi Myopathy, are linked to mutations in dysferlin). Using mutagenesis and deletion strategies, we are identifying the regions of the molecule responsible for targeting the protein to the TT, and the regions responsible for activity in vitro. The figure, below, shows a fluorescently tagged version of wild type dysferlin and a mutant. The WT protein targets the t-tubules normally (note the doublet of label, expected of t-tubule proteins) but the mutant does not, and instead concentrates in membranes around the ends of each sarcomere. (NB: the large oval gaps are nuclei.)

Parallel studies are underway to pinpoint the role of dysferlin in stabilizing the TT in the muscles of mice subjected to large strain injuries from lengthening contractions in vivo. A key goal of all these studies is to identify small molecules with potential therapeutic value in dysferlinopathic mice, for use as possible therapies for LGMD2B and MM in man.
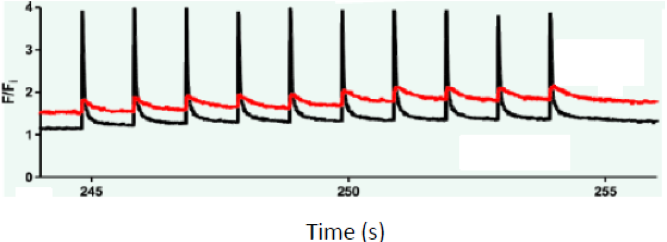
In one series of experiments, completed recently, we showed that the control of Ca2 fluxes and baseline Ca2+ concentrations in the myoplasm of dysferlin-null muscle fibers is much more easily disrupted than in controls. Visualizing the intracellular Ca2+ with a fluorescent dye and quantitating the fluorescent signal, we found that a brief osmotic shock to dysferlin-null fibers, but not to controls, greatly reduced the size of the Ca2+ transient elicited by an action potential, and that this change was accompanied by a significant and prolonged increase in the baseline levels of Ca2+ (see figure; red = dysferlin-null; black = WT). This increase in Ca2+ is likely to induce a change in the mode of Ca2+ release from the terminal cisternae of the sarcoplasmic reticulum, from voltage-induced to Ca2+ induced. Ca2+-induced Ca2+ release in skeletal muscle is associated with pathology and may well underlie the pathogenesis of dysferlinopathies.
In our most recent studies, we have begun to dissect the functions of the different domains of dysferlin (it has 7 C2 domains and several ferlin domains) in regulating Ca2+ signaling in skeletal muscle. Our results to date suggest that different C2 domains influence the Ca2+ release mechanism in distinct ways.