Molecular Mechanisms of Muscular Dystrophies
A major focus of many of our studies has been to learn how changes in the composition and organization of the membrane systems of skeletal muscle can lead to muscular dystrophy. We have learned that the organization of structural proteins at the sarcolemma, and especially at costameres, is affected in several forms of muscular dystrophy. The changes at costameres are likely to account in part for the increased susceptibility to injury and to muscle weakness, both of which are typical of dystrophic muscle.
Our earlier studies to date have focused on an animal model for Duchenne Muscular Dystrophy and for a congenital muscular dystrophy. We are now focusing on two other forms of muscular dystrophy, Facioscapulohumeral Muscular Dystrophy (FSHD), and the dysferlinopathies, Limb Girdle Muscular Dystrophy Type 2 B and Miyoshi Myopathy (LGMD2B/MMD1).
FSHD
FSHD is an autosomal dominant disease that affects 1 in 8,000 Americans. We are using our xenografting methods to create grafts of FSHD muscle tissue, to study the pathogenesis of the disease and to determine the physiological, morphological and biochemical changes that underlie it. (Please see the previous section for details.)
We have also used large format 2D gel electrophoresis in a custom-built chamber (see figure) to characterize the proteome of muscles from patients with FSHD, to identify the proteins responsible for the altered costameres. The gels, which we typically analyze in quadrants (see figure) resolve ~2000 spots reliably.


Mu-Crystallin in Muscle and Muscle Disease
To date, we have identified several proteins that change in FSHD muscle, and characterized one: µ-crystallin. µ-Crystallin is a thyroid-hormone binding protein present in the cytoplasm of many mammalian that regulates gene expression by controlling the amount of free thyroid hormone via the thyroid hormone receptor. Work in progress suggests that μ-crystallin alters the transcription of muscle-specific thyroid hormone-dependent genes and shifts metabolism such that muscle utilizes more fat as an energy source. High levels of µ-crystallin expression also block muscle development in culture (figure). Current studies address the variation of protein levels by family and state of health by examining muscle biopsies and metabolic testing. We have also created a transgenic mouse that overexpresses Crym in its skeletal muscle, to learn how it regulates gene expression and metabolism and if it can affect the pathologies associated with muscular dystrophy

LGMD2B/MMD1
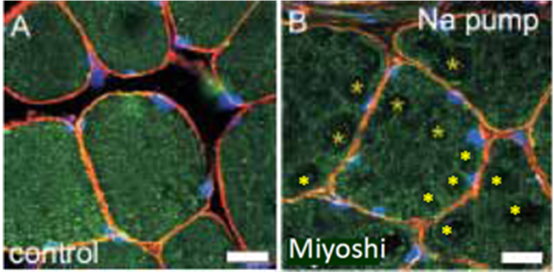
Our studies of LGMD2B/MMD1 are primarily carried out in mice that carry similar mutations to those that cause the human disease. Whenever possible, we examine human muscle tissue to ensure that the results we obtain in mice are congruent with findings in man. Typically, this involves examining biopsies of human muscles. As mentioned above, the absence of dysferlin results in unstable transverse tubules and misregulation of Ca2+. To learn if the transverse tubules of muscle from dysferlinopathic patients are also unstable, we immunolabeled frozen sections of muscle biopsies for a marker protein, the Na,K-ATPase (α2 isoform). As shown below, the integrity of the transverse tubules is severely compromised in muscle from an MMD1 patient (Note the gaps, labeled with yellow asterisks). This and related findings support the idea that, as in mice and in our in vitro studies, LGMD2B/MMD1 in many is a disease of the transverse tubules that results in misregulation of Ca2+ signaling